TopHat2 uses using Bowtie to align RNA-Seq reads to mammalian-sized genomes and then analyzes the mapping results to identify splice junctions between exons. This step is optional.

Tophat Cufflinks Command Pipeline
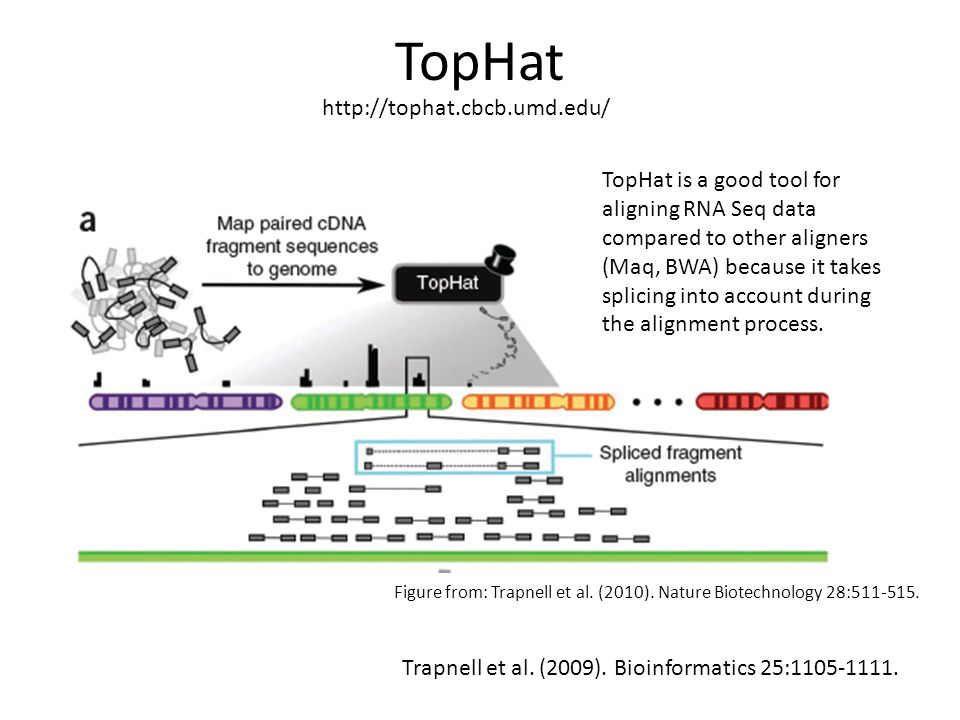
TopHat is a fast splice junction mapper for RNA-Seq reads.
. STAR GSNAP and MapSplice. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Tophat is one of several aligners developed specifically for RNA-seq eg.
This is quite different conceptually to mapping to the transcriptome directly. The protocol assumes that RNASeq was done using Illumina or Solid sequencing techniques. Transcriptome analysis via RNA-Seq.
Another reason to use it is that it works well together with Cufflinks. This tutorial will focus on doing a 2 condition 1 replicate transcriptome analysis in mouse. Using TophatCufflinksedgeR to analyze RNAseq data Step 1.
Both Tophat and Cufflinks require a reference genome. A_s_2_sequencetxt tophat_1log Step 4. Another vignette Di erential analysis of count data the DESeq2 package covers more of the advanced details at a faster pace.
There are several types of RNA-Seq. Nature Protocols serial online. Matrices from raw sequencing data.
We recommend that you watch the video Aligning RNA-seq reads to reference genome instead which covers t. Since your reads came from spliced transcripts in an RNA-Seq experiment Bowtie will identify islands in your reference genomewhere. TopHat will map your reads first by running Bowtie to identify places where reads map end to end.
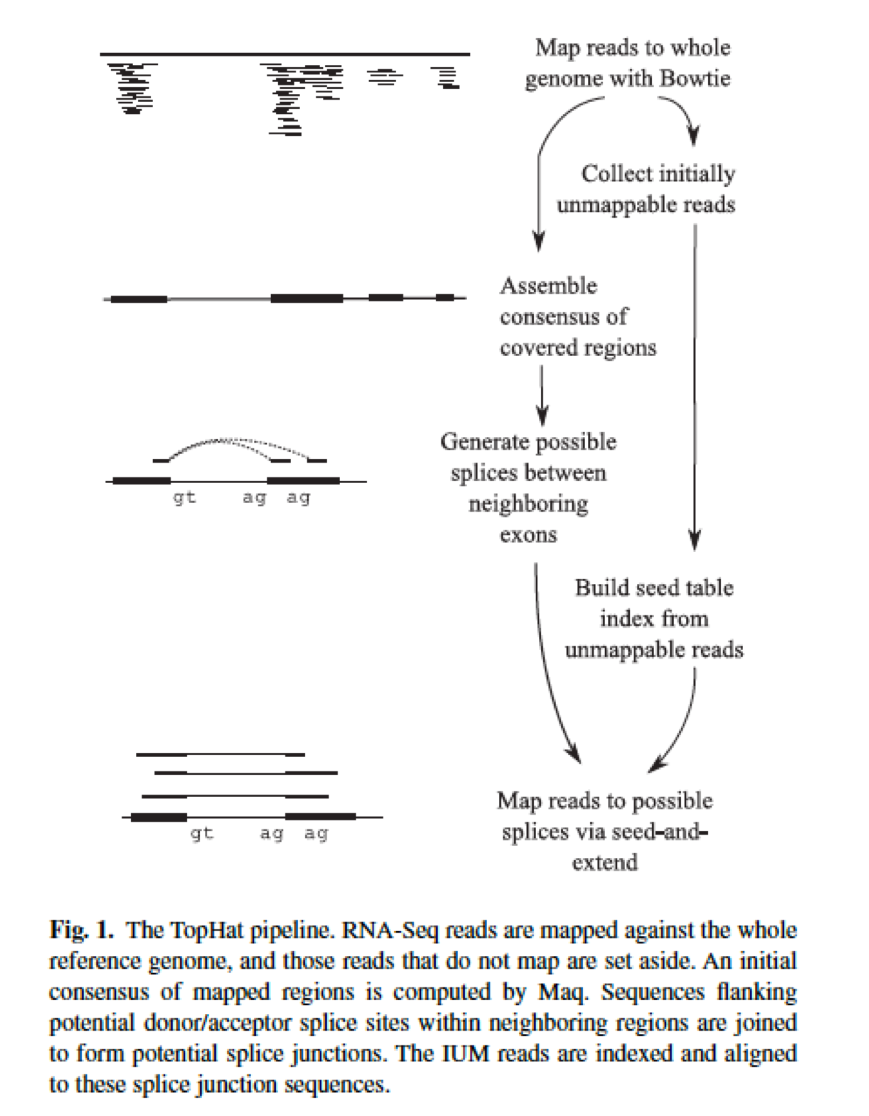
Using this initial mapping information TopHat builds a database of possible splice junctions and then maps the reads against these junctions to confirm them. This tutorial is inspired by an exceptional RNA seq course at the Weill Cornell Medical College compiled by Friederike Dündar Luce Skrabanek and Paul Zumbo and by tutorials produced by Björn Grüning bgruening for Freiburg Galaxy instance. Reference-Based RNA-Seq Data Analysis Workshop Session 2 Exercise.
These FASTQ files are RNA-seq data from two samples. Much of Galaxy-related features described in this section have been developed by. Using Samtools to index the bam file then visualize the alignment using IGV.
It aligns RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short read aligner Bowtie included in this plugin and then analyzes the mapping results to identify splice junctions between exonsThis plugin runs on Mac OS and 64-bit Linux only it is not supported Windows. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks Trapnell et al 2012. The real RNA-seq data would normally take hours to process.
This vignette is designed for users who are perhaps new to analyzing RNA-Seq or high-throughput sequencing data in R and so goes at a slower pace explaining each step in detail. Transcriptome splice-variantTSSUTR analysis microRNA-Seq etc. As every sequence record takes up 4 lines in the fastq file the line number divided by 4 gives you the number of sequencing reads in the file.
First we will map the reads to a reference genome using TOPHAT. RNA-Seq Tutorials Tutorial 1 RNA-Seq experiment design and analysis Instruction on individual software will be provided in other tutorials Tutorial 2 Advanced RNA-Seq Analysis topics Hands-on tutorials Analyzing human and potato RNA-Seq data using Tophat and Cufflinks in Galaxy. The experiment and analysis protocol we will follow is derived from a paper in Nature Protocols by the research group responsible for one of the most widely used set of RNA-seq analysis tools.
It is for visualizing the read alignment to the genome. Bowtie1 is an ultrafast memory-effi cient aligner for large sets of short reads. RNA-Seq Tutorials Tutorial 1 RNA-Seq experiment design and analysis Instruction on individual software will be provided in other tutorials Tutorial 2 Hands-on using TopHat and Cufflinks in Galaxy Tutorial 3 Advanced RNA-Seq Analysis topics.
This tutorial from 2017 covers the TopHat aligner. It is not necessarily the best one in terms of speed and performance but it is actively improved and maintained and has a large user base. By first mapping RNA-Seq reads to the genome TopHat identifies potential exons since many RNA-Seq reads will contiguously align to the genome.
Some of the applications used in RNA sequencing analysis are the following. Tophat is a splicing aware aligner so we can map transcripts to the genome. References Trapnell C Roberts A Pachter L et al.
Mapping RNA-seq reads to the genome with tophat In this tutorial well map reads from an RNA-seq study in Drosophila melanogaster to the reference genome using tophat.
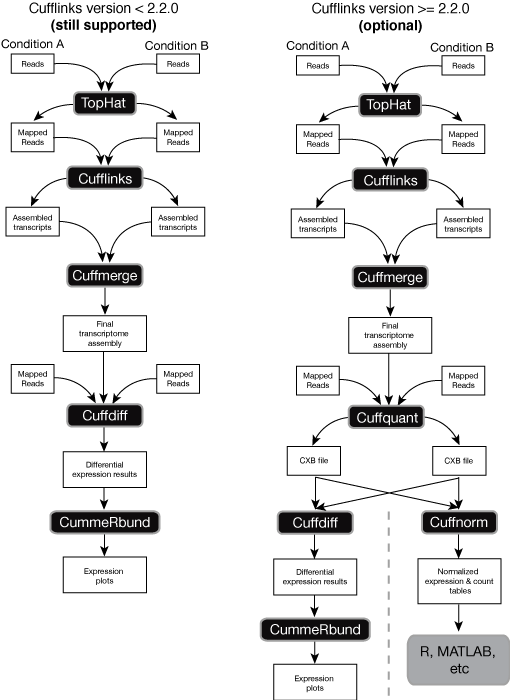
The Cufflinks Rna Seq Workflow

Reference Based Rnaseq Data Analysis Long
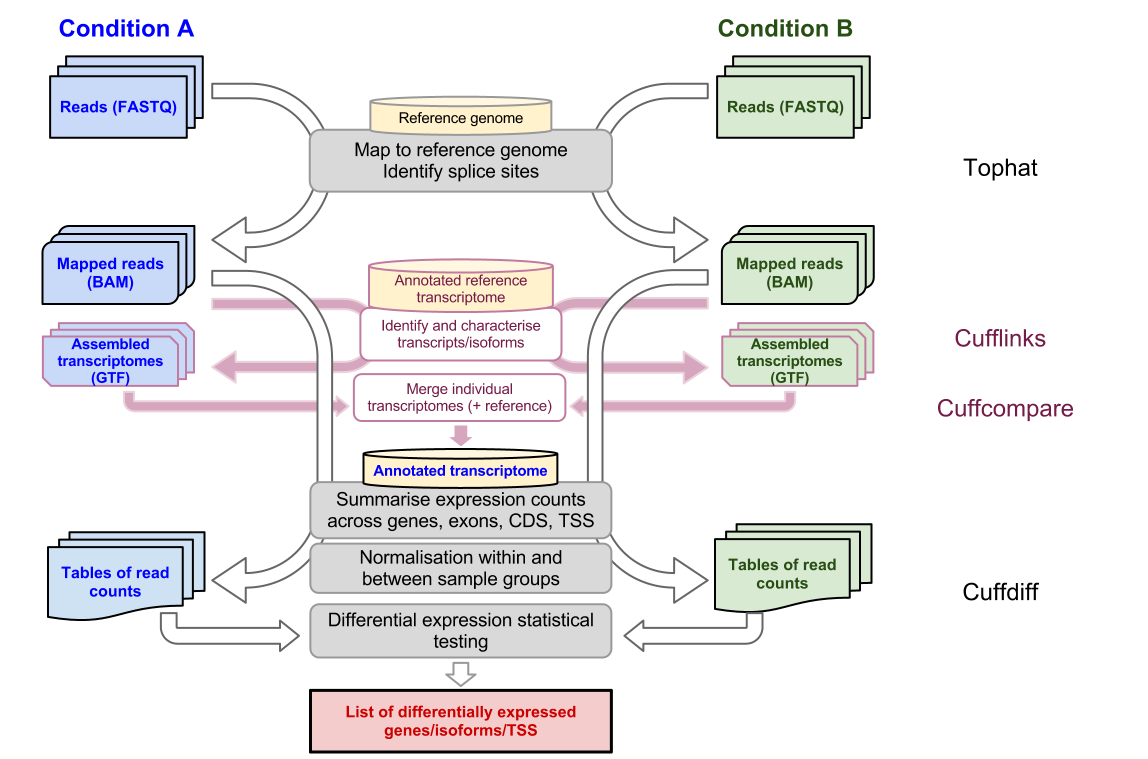
Introduction To Bulk Rnaseq Analysis Bioinformatics Documentation

Basic Analyses With Tophat Cufflinks Rnaseq Tutorial 1 Documentation

Rna Seq Alignment And Visualization Youtube
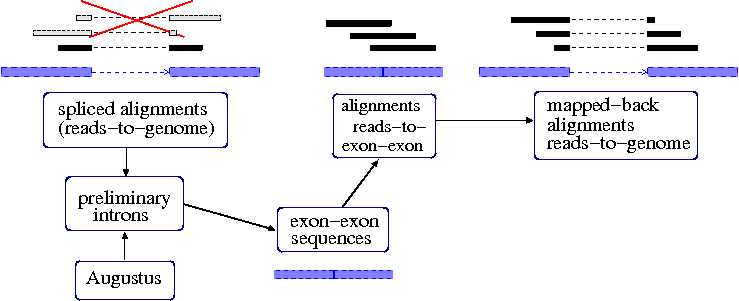
Incorporating Rnaseq Tophat To Augustus Computational Biology
Imgs 2012 Bioinformatics Workshop Rna Seq Using Galaxy Ppt Video Online Download
Aligning Rna Seq Data Ngs Analysis
0 comments
Post a Comment